





Smart-Seq2
上一节我们介绍了10x Genomics的原理及分析方法。Smart-seq2是一种单细胞RNA测序技术,旨在对单个细胞的基因表达进行高分辨率的分析。以下是Smart-seq2的发展历史和技术特点的详细介绍:
发展历史:
Smart-seq: Smart-seq2的前身是Smart-seq,它是首个用于单细胞转录组测序的方法。Smart-seq的提出可以追溯到2012年,它的原理是通过在反转录过程中引入一个定制引物,将细胞内的RNA转录本进行全长合成。这使得可以获得单细胞水平上更详细和全面的基因表达数据。
Smart-seq2的改进: 随着技术的发展,Smart-seq2在Smart-seq的基础上进行了改进,以提高单细胞RNA测序的效率和覆盖度。Smart-seq2的引物设计更加精确,可以更好地应对低质量RNA样本,并且在捕获基因表达信息时表现更为灵敏。

技术特点:
1.全长转录本测序: Smart-seq2使用全长转录本测序的方法,使得可以获取细胞中每个基因的完整转录本信息,而不是只关注特定区域。这有助于更准确地了解单细胞的基因表达谱。
2.高灵敏度: 引物设计的改进和全长转录本测序的特点使得Smart-seq2在捕获低表达基因和检测单个细胞中稀有转录本方面更为灵敏。
3.适用性广泛: Smart-seq2的技术特点使其适用于各种细胞类型和样本来源,包括低质量的单细胞RNA。
4.单细胞分辨率: Smart-seq2具有较高的单细胞分辨率,可以在单细胞水平上深入挖掘细胞异质性和亚型。
5.低输入RNA样本: 与一些其他单细胞RNA测序方法相比,Smart-seq2对RNA输入量的要求较低,这使得可以应用于限制性的细胞样本。
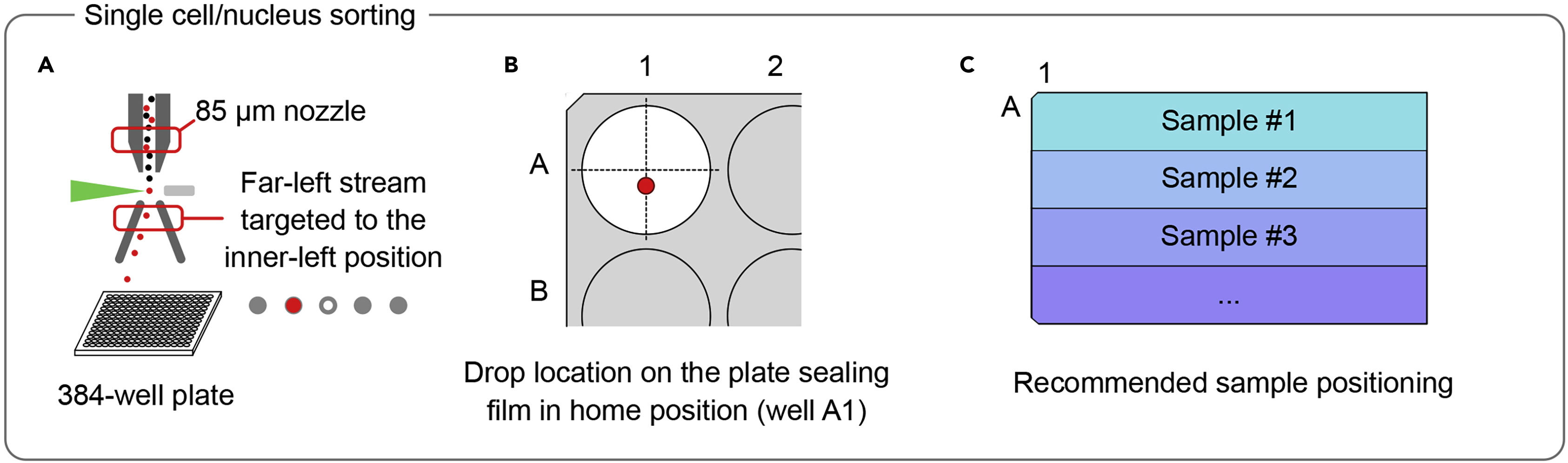
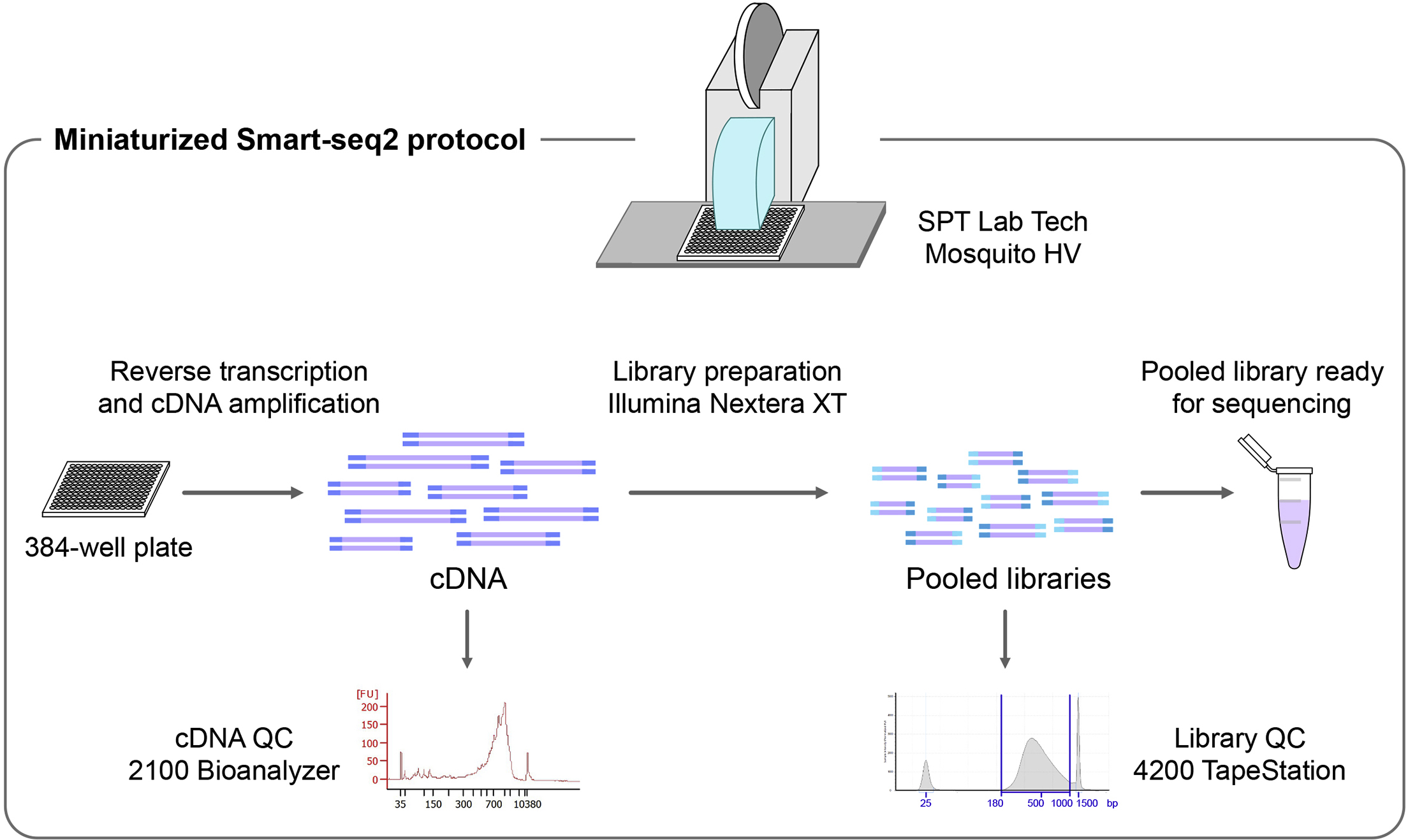
分析流程:
和10x Gemonics不同,Smart-seq2在建库过程中,对每个孔中添加的index序列是已知的,所以整体分析思路是首先根据每个孔板中的index拆分fastq,然后进行比较、定量、基因表达技术。流程基本和bulk RNA-seq分析流程相似。最后将每个孔板中基因表达水平合并为所有细胞的表达。主要包括以下步骤:
1.质控
使用fastqc软件对原始测序数据进行质控,fastqc的使用见[教程](),(待补充)
2.拆分
sabre是一款根据barcode(index)进行拆分的软件,在github上下载之后(如果无法下载,请右侧扫码联系博主),执行下述命令
$ cd /path/to/sabre
$ make执行完成之后,用ls查看当前目录,新生成的sabre即可使用
$ ls -l .
$ ll
total 67
-rw-r--r-- 1 *** group 1023 Feb 3 2021 LICENSE
-rw-r--r-- 1 *** group 924 Feb 3 2021 Makefile
-rw-r--r-- 1 *** group 4160 Feb 3 2021 README.md
-rw-r--r-- 1 *** group 1312 Feb 3 2021 barcode.o
-rw-r--r-- 1 *** group 16392 Feb 3 2021 demulti_paired.o
-rw-r--r-- 1 *** group 12104 Feb 3 2021 demulti_single.o
-rwxr-xr-x 1 *** group 27183 Feb 3 2021 sabre #可执行文件
-rw-r--r-- 1 *** group 3424 Feb 3 2021 sabre.o
drwxr-xr-x 2 *** group 4096 Feb 3 2021 src
sabre可用于拆分单端和双端测序数据,具体用法如下:
$ sabre
Usage: sabre <command> [options]
Command:
pe paired-end barcode de-multiplexing
se single-end barcode de-multiplexing
--help, display this help and exit
--version, output version information and exit
我们这里都双端测序结果进行拆分,使用方法如下:
$ sabre pe \
-r R1.fq.gz \ #R1
-f R2.fq.gz \ #R2
-b barcode.txt \ #barcode: barcode_seq file1_out file2_out
-m 2 \
-u unknown_sepID.fastq \
-w unknown_UMI.fastqaddUMItoSeqID
自己编程将序列中的UMI截取下来添加至SeqID中。我是用python3编写的,代码如下
import os
import sys
import argparse
import logging
import gzip
def main():
uopen = gzip.open if args.umi_fq.endswith('gz') else open
sopen = gzip.open if args.seq_fq.endswith('gz') else open
with uopen( args.umi_fq ) as umi, uopen( args.seq_fq ) as seq, open( args.out_fq, "w" ) as out:
for reads_ID in umi:
umi_seq = umi.readline()[0:args.umi_length];
umi.readline();
umi.readline()
seq.readline()
reads_seq = seq.readline()
seq.readline()
quality = seq.readline()
reads_ID = reads_ID.split()
if args.umi_fq.endswith('gz'):
reads_ID = [ bytes.decode(i) for i in reads_ID ]
if args.seq_fq.endswith('gz'):
umi_seq = bytes.decode(umi_seq)
reads_seq = bytes.decode(reads_seq)
quality = bytes.decode(quality)
reads_ID = "".join( [ reads_ID[0], "+UMI_", umi_seq, " ", reads_ID[1] ] )
out.write( reads_ID + "\n" + reads_seq + "+\n" + quality )
if __name__=="__main__":
main()之后使用cutadapt和trimmomatic过滤接头序列以及低质量序列
比对
使用STAR进行比对,featureCounts进行定量,此部分步骤可以参考bulk RNA-seq分析。最终获得每个基因的唯一UMI数量作为基因的表达量
表达矩阵
将所有细胞的表达量合并成为表达矩阵进行下游分析。
参考文献
除非注明,文章均为原创,转载请以链接形式标注本文地址
本文地址:http://bioschool.cn/omics/scrna/smart-seq2/