





单细胞多组学及分析方法
2020年,Daehee Hwang团队在《Experimental & Molecular Medicine》(IF≈8.7)杂志上发表了一篇关于单细胞技术及分析方法的综述。通过这篇综述,我们可以从整体上学习单细胞多组学的应用。
在过去的十年中,基于NGS技术,我们对癌症的发展有了深刻的认识,但这是针对上百万个细胞进行的研究,而癌症的异质性在这个过程中是被忽略或者低估的。单细胞,从字面上即可理解通过从实体瘤或者血液肿瘤中解离细胞并进行分离,提取细胞中的DNA、RNA或者蛋白质等信息进行分析,实现在单个细胞水平上刻画每个细胞的特征。
多组学的分析有助于更加全面的了解细胞,其主要特点包括:1)单细胞分离、UMI及测序技术可以从单个细胞中获得多种类型的分子2)对分子的整合分析可以用于表征细胞类型及其与病理生理过程相关的功能。
细胞分离和编码
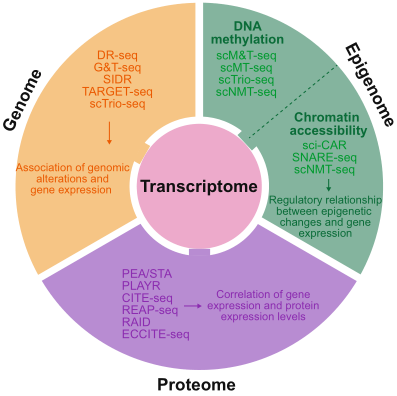
单细胞多组学分析的第一步需要从同一细胞中分离出多种类型的分子,包括分离单个细胞及对多种类型的分子进行编码。可以通过不同的方法捕获单细胞,如显微解剖、细胞分选、微流控平台等。捕获单个细胞之后可以通过离心的方法将细胞核从细胞质中分离并获得gDNA,而mRNA可以使用oligo-dT分离得到,分离之后对DNA和RNA进行编码,这一方法易导致DNA或者mRNA丢失。在另外一种方法中,裂解液处理之后不进行mRNA和DNA的分离而直接进行逆转录,将mRNA转为单链cDNA,然后对gRNA和cDNA进行扩增,并将扩增产物分为两部分,利用PCR对其中一半扩增出gDNA,另一半扩增出cDNA。
单细胞基因组和转录组数据的整合分析
单细胞基因组测序方法包括MDA、MALBAC、PicoPLEX等。而单细胞转录组技术包括Quartz-seq、Smart-seq、CEL-seq等。同时进行单细胞基因组和转录组的方法包括scTrio-seq 、G&T-seq、DR-seq、SIDR及TARGET-seq。
转录组和表观组的整合分析
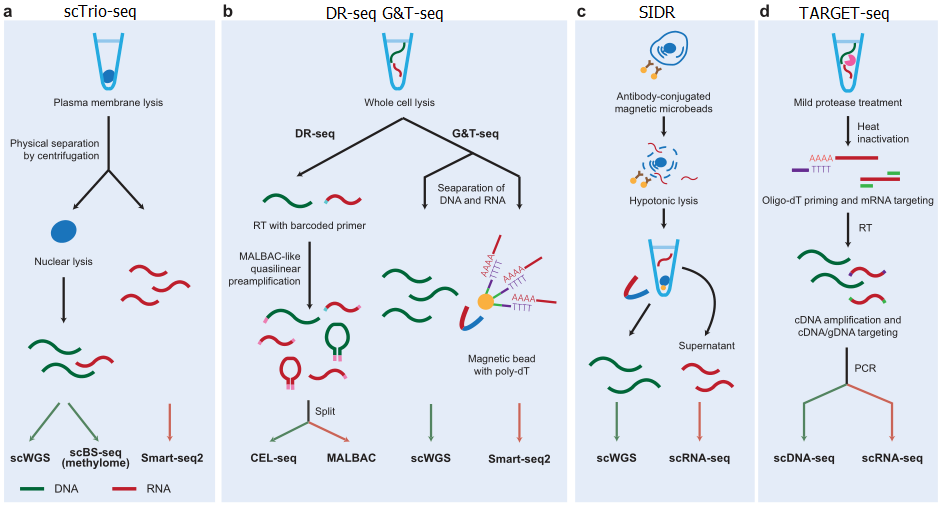
DNA甲基化、组蛋白修饰(甲基化与乙酰化)、染色质开放性对基因的表达都有重要影响,且已经实现在单细胞水平衡量表观水平。用于检测DNA甲基化的单细胞亚硫酸盐测序方法包括scRRBS、scWGBS、snmC-seq、sci-MET。第一个单细胞表观和转录组分析方法为scM&T-seq,是基于G&T-seq方法改进而来。scMT-seq使用微管从细胞中吸取细胞核并分别进行scRRBS和Smart-seq2测序。scTrio-seq可以同时实现基因组、甲基化及转录组测序。
染色质免疫共沉淀技术可以用于衡量单个细胞水平上的组蛋白修饰。而染色质开放性研究的方法包括scDNase-seq、sci-ATAC-seq、scATAC-seq、NOMe-seq、scMNase-seq。基于这些方法,染色质开放性和转录组整合分析的方法包括sci-CAR、SNARE-seq、scNMT-seq。
转录组和蛋白组的整合分析
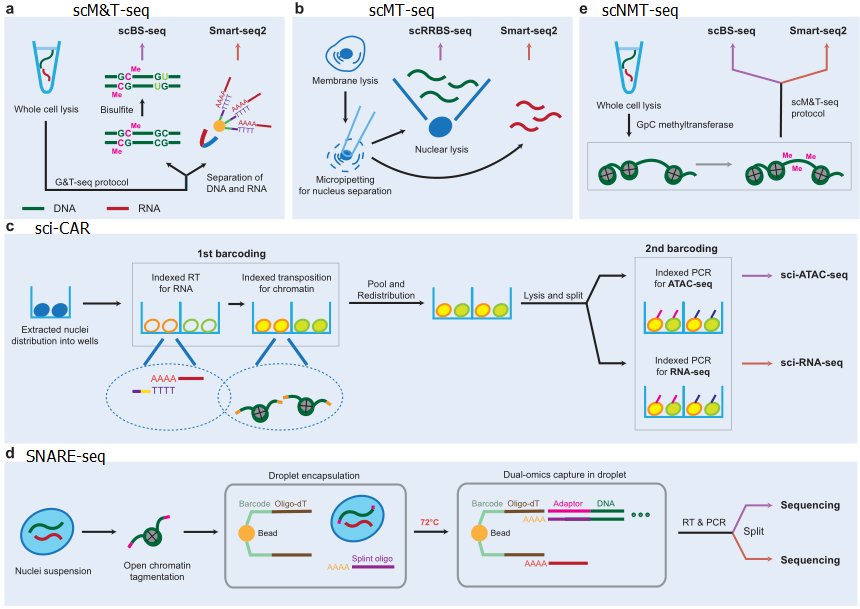
尽管蛋白质特别重要,但是受限于蛋白无法扩增,导致单细胞水平可测量的蛋白很少。目前同时测量转录组和蛋白组的方法有PEA/STA、PLAYR、CITE-seq以及REAP-seq。CITE-seq和REAP-seq通过使用抗体标记细胞表面marker来鉴定细胞表面蛋白。而RAID技术可以检测细胞内部的蛋白或者磷酸化的蛋白。
单细胞组学数据分析
从单细胞单组学数据中我们可以获得单细胞水平不同类型的信息,如突变及CNV等基因组变异、DNA甲基化位点、染色质开放区域以及mRNA和蛋白异常。对于scRNA-seq数据,PCA(主成分分析)、tSNE被广泛用于数据降维。聚类方法则在距离计算、聚类算法和对基因和细胞同时聚类的能力有所不同。分析方法包括Seurat、pcaReduce、SC3、BackSPIN、SNN-cliq。基于单细胞数据推断调控网络的方法和普通转录组数据不同,目前开发出的方法包括SCNS、inferenceSnapshot、SCODE、SCENIC。Monocle、DPT、Wishbone、Waddington-OT方法则可以用来推断细胞轨迹。scWGS的主要目的在于获得单细胞水平的拷贝数变异及识别SNV等信息。可以单细胞基因组数据中获取CNV信息的方法包括Ginkgo、baseqCNV、SCNV、SCCNV和SCOPE。SNV的分析软件有SCcaller, baseqSNV, MonoVar和SCAN-SNV。单细胞表观组主要用来识别染色质开放区域及识别DNA甲基化位点。由于低覆盖度的特点,很难识别开放区域或者甲基化峰。一个可行的方案为汇总大约100个细胞的数据之后,识别出包含的峰的位置再在单个细胞中分析是否包含这些峰。scABC采用这个方案从scATAC-seq数据中识别染色质开放区域。
单细胞多组学数据整合分析
单细胞多组学数据的整合分析策略主要包括单组学数据之间的相关性分析、将某一组学数据合并至另一组学数据中以及整合所有的组学数据生成合并后的单细胞谱。在第二种策略中,scRNA-seq数据是最常被合并的组学数据,因为转录组数据和其他类型数据相比,覆盖度更高。针对第三种策略,已经有几种基于矩阵的方法开发出来,如LIGER、MOFA。
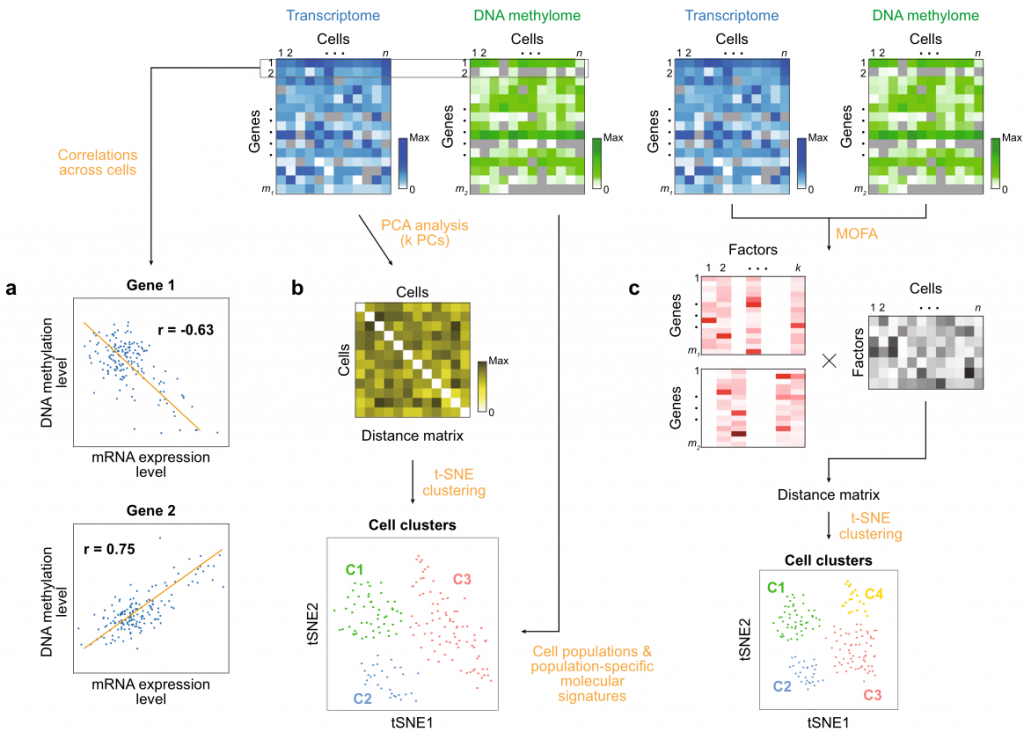
和单细胞单组学数据相比,多组学数据可以通过更全面的描述来系统解释细胞的多样性和异质性。多组学数据在解释细胞内调控因果关系时具有优势,如基于单细胞基因组和转录组数据可以分析基因组突变对转录组的影响,单细胞表观组和转录组数据的分析可以揭示表观改变和基因表达之间的关系。
除非注明,文章均为原创,转载请以链接形式标注本文地址
本文地址:http://bioschool.cn/omics/sc-multi-omics/%e5%8d%95%e7%bb%86%e8%83%9e%e5%a4%9a%e7%bb%84%e5%ad%a6%e5%8f%8a%e5%88%86%e6%9e%90%e6%96%b9%e6%b3%95/